
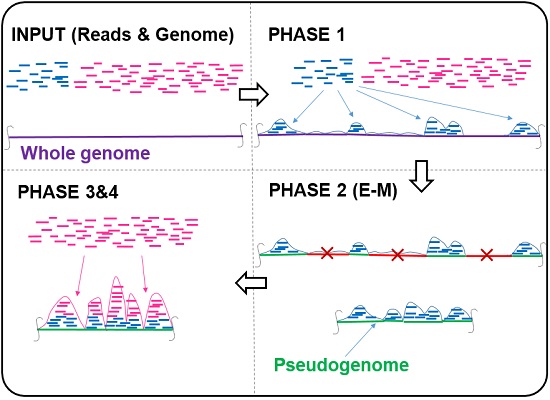
Map2Peak: From Unmapped Reads to ChIP-Seq Peaks in Half the Time
The standard ChIP-Seq data analysis workfow to identify transcription factor binding sites consists of two separate stages, i.e., read mapping and peak calling, performed in tandem. All raw (unmapped) reads are aligned to the reference genome using a read mapping tool, and then the mapped read dataset is fed into a peak caller. Despite the advances in read mapping and peak calling tools, this standard tandem workfow has inherent major drawbacks: (1) Since the majority of reads in ChIP-Seq datasets are background reads, the read mapper spends the majority of its computational time on mapping reads that have no role in forming the true peaks. (2) The overwhelming presence of background reads in the mapped read dataset results in calling many false peaks. This is sometimes addressed by using control datasets at the cost of adding to the computational burden of the work ow even more. (3) Multi-mappable reads (reads that align to more than one location on the genome) are often discarded at the read mapping stage to avoid false peaks, resulting in the inability of the standard workflow to identify true peaks in important repeat elements of the genome. This is while most read location resolver tools are computationally expensive. We present Map2Peak, a novel tool aimed at eliminating the aforementioned drawbacks. Map2Peak receives ChIP-Seq unmapped read dataset as the input and presents the peaks as the output at a speed twice faster than that of standard tandem workflow (Bowtie + MACS2) but with comparable accuracy. Contrary to the standard workflow, Map2Peak intertwines partial read mappings, a statistical model to filter the genome, and peak calling in a four-phase algorithm. Map2Peak uses the fragment count pro le information obtained during the early stages of read mapping (Phase 1) to capture the fragment coverage as a 2-component Poisson mixture model, which is then used in an expectation-maximization (E-M) approach to divide the genome into candidate background and enriched regions (Phase 2). The remaining reads are then restricted to be mapped exactly only to the much shorter pseudo-genome composed of the candidate enriched regions (Phase 3), resulting in a signifi cant speed-up (up to 2 times). Peak calling is then performed on the pseudo-genome (Phase 4). Our computational results show that the peaks called by Map2Peak encompass most of the peaks called by the standard workflow (85%-96%) and many novel motif-justi able peaks which are not detected by the standard work ow due to the background noise. The majority of background reads do not align to the candidate enriched regions in Phase 3, resulting in Map2Peak correctly discarding at least 85% of the background reads. This signi cantly alleviates the need for control datasets. Moreover, Map2Peak does not discard multi-mappable reads as it implicitly resolves the location of the ones that uniquely map to the pseudo-genome. Consequently, about 50% of the novel peaks called by Map2Peak are in the repeat elements of the genome, much higher than the percentage of non-novel peaks on these elements. Although Map2Peak does not resolve the location of all multi-mappable reads but does so for those which are more likely to form a peak, and hence, serves the same purpose as incorporating read location resolver tools into the standard workflow.
FAMOUS: Fast Approximate string Matching using OptimUm search Schemes
Finding approximate occurrences of a pattern in a text using a full-text index is a central problem in bioinformatics. Bidirectional indices have opened new possibilities for solving the problem as they allow the search to be started from anywhere within the pattern and extended in both directions. In particular, use of search schemes (partitioning the pattern into several pieces and searching the pieces in certain orders with bounds on the number of errors in each piece) has shown significant potential in speeding up approximate matching. However, finding the optimal search scheme to maximize the search speed is a difficult combinatorial optimization problem. In this paper, we propose, for the first time, a method to solve the optimal search scheme problem for Hamming distance with given number of pieces. Our method is based on formulating the problem as a mixed integer program (MIP). We show that the optimal solutions found by our MIP significantly improve upon previously published ad-hoc solutions. Our MIP can solve problems of considerable size to optimality in reasonable time and has the attractive property of finding near-optimal solutions for much larger problems in a very short amount of time. In addition, we present FAMOUS (Fast Approximate string Matching using OptimUm search Schemes), a bidirectional search (for Hamming and edit distance) implemented in SeqAn that performs the search based on the optimal search schemes from our MIP. We show that FAMOUS is up to 35 times faster than standard backtracking and anticipate that it will improve many tools as a new core component for approximate matching and NGS data analysis. We exemplify this by searching Illumina reads completely in our index at a speed comparable to or faster than current read mapping tools. Finally, we pose several open problems regarding our MIP formulation and use of its solutions in bidirectional search.

2018
Kianfar, Kiavash; Pockrandt, Christopher; Torkamandi, Bahman; Luo, Haochen; Reinert, Knut
Optimum Search Schemes for Approximate String Matching Using Bidirectional FM-Index Journal Article
In: In review, 2018.
@article{Kianfar2018,
title = {Optimum Search Schemes for Approximate String Matching Using Bidirectional FM-Index},
author = {Kiavash Kianfar and Christopher Pockrandt and Bahman Torkamandi and Haochen Luo and Knut Reinert},
url = {https://arxiv.org/abs/1711.02035v2},
year = {2018},
date = {2018-01-01},
journal = {In review},
abstract = {Finding approximate occurrences of a pattern in a text using a full-text index is a central problem in bioinformatics and has been extensively researched. Bidirectional indices have opened new possibilities in this regard allowing the search to start from anywhere within the pattern and extend in both directions. In particular, use of search schemes (partitioning the pattern and searching the pieces in certain orders with given bounds on errors) can yield significant speed-ups. However, finding optimal search schemes is a difficult combinatorial optimization problem.
Here for the first time, we propose a mixed integer program (MIP) capable to solve this optimization problem for Hamming distance with given number of pieces. Our experiments show that the optimal search schemes found by our MIP significantly improve the performance of search in bidirectional FM-index upon previous ad-hoc solutions. For example, approximate matching of 101-bp Illumina reads (with two errors) becomes 35 times faster than standard backtracking. Moreover, despite being performed purely in the index, the running time of search using our optimal schemes (for up to two errors) is comparable to the best state-of-the-art aligners, which benefit from combining search in index with in-text verification using dynamic programming. As a result, we anticipate a full-fledged aligner that employs an intelligent combination of search in the bidirectional FM-index using our optimal search schemes and in-text verification using dynamic programming outperforms today's best aligners. The development of such an aligner, called FAMOUS (Fast Approximate string Matching using OptimUm search Schemes), is ongoing as our future work.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Here for the first time, we propose a mixed integer program (MIP) capable to solve this optimization problem for Hamming distance with given number of pieces. Our experiments show that the optimal search schemes found by our MIP significantly improve the performance of search in bidirectional FM-index upon previous ad-hoc solutions. For example, approximate matching of 101-bp Illumina reads (with two errors) becomes 35 times faster than standard backtracking. Moreover, despite being performed purely in the index, the running time of search using our optimal schemes (for up to two errors) is comparable to the best state-of-the-art aligners, which benefit from combining search in index with in-text verification using dynamic programming. As a result, we anticipate a full-fledged aligner that employs an intelligent combination of search in the bidirectional FM-index using our optimal search schemes and in-text verification using dynamic programming outperforms today's best aligners. The development of such an aligner, called FAMOUS (Fast Approximate string Matching using OptimUm search Schemes), is ongoing as our future work.